All medical device companies need to include a success rate and time to decide on their submissions in their go-to-market plan. It turns out that helpful information on both subjects is readily available from the FDA. In this article, I share my analysis of different data sources to provide answers to two questions:
1. What is the success rate for medical device submissions?”
2. How long will it take for the FDA to give us a decision on our medical device submission?
A few weeks ago, an early-stage founder with a novel medical device asked me, “What is the success rate for FDA medical device submissions?” This excellent question prompted me to ask, “What’s a typical Turnaround Time (TaT) for a decision?” Answering these questions took me down an exciting path, and I’m sharing those answers with you in this article. I’m focused on 510(k) and original PMA submissions, but data is available for other submission types, as you will see below.
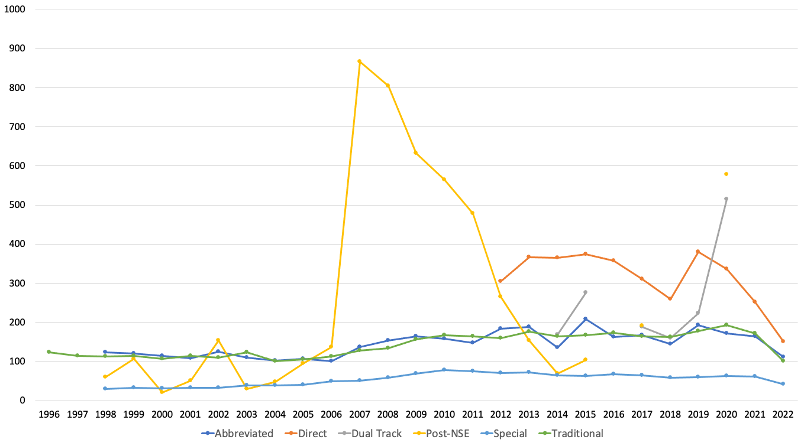
My first stop was the FDA’s primary website for medical devices. While finding what you want on this page is not always easy, it’s an excellent first stop for any search. I start by poking around the site to find keywords which I then put into a commercial search engine. (Note: The FDA launched a revamped search tool while I was working on this article. Unfortunately, this tool does not produce better results than the original search bar.) After some browsing, I found the device Downloadable 510(k) Files page, under Device Approvals, Denials, and Clearances. These files list the summaries or statements for final 510(k) decisions made during specific periods. I downloaded the 1996-current file and opened it in Excel. After reviewing the file layout, adding a couple of calculated fields, and creating a few pivot tables and charts, I had an answer to the time question (Figure 1). Discontinuous lines indicate no data in the table for the corresponding years. From this data, we can see that the average time to decision varies quite a bit between different types of 510(k) submissions. The standard deviations are relatively large, too, ranging from 66 days for Special and 540 days for post-NSE types. The most common types, Traditional and Abbreviated, take an average of 140 days (4.7 months) for a decision. A downward trend in average TaT appears to have started in 2018. This trend may reflect the performance improvement plan prompted by the Medical Device User Fee Act (MDUFA) renewal in 2017 (MDUFA IV).
Next, I asked if similar data is available for Pre-Market Applications (PMAs). These are the submissions required for Category III devices with the highest level of risk. Back at the FDA website, the PMA Approvals page had the link for the downloadable file containing data for the releasable PMAs and their associated file description. There is just one file containing original and supplemental PMAs of all types from the 5th of each month backward to 1990. Once again, I opened this file in Excel, added a couple of calculated fields, and created some pivot tables and charts. Figure 2 shows the results for original PMAs plus two types of supplements, 180-day, and Panel Track. It’s not surprising that original PMAs take, on average, longer than supplemental PMAs. Originals have an average TaT of 449 days, Panel track of 327 days, and Normal 180-day track of 204 days. The standard deviations are 368, 342, and 176 for Original, Panel, and Normal, respectively. The downward trend in average TaT in the 510(k) data is also observed for PMAs. Both the 510(k) and PMA data show you can expect to wait a while for a decision on your submission, and that wait time can vary quite a lot.

[FYI, different types of supplements are required for approved devices based on the level of change and how it may impact the safety or effectiveness of the devices. Interested readers may find out more about PMA supplements here. For 510(k)s, changes are evaluated based on the original device acting as the predicate. Specific changes are ordinarily filed as a Special 510(k). Sounds complicated? In some ways, it is. Current guidance for the program is here.]
You’ll recall I mentioned MDUFA IV earlier. This acronym is the 2017 version of the Medical Device User Fee Amendments to the Federal Food, Drug, and Cosmetic Act. These amendments, first authorized in 2002, allow the FDA to collect user fees for the review of medical device submissions. The user fees are used to improve the efficiency of regulatory processes to encourage innovation and increase access to safe and effective medical devices. Similar programs exist for prescription drugs and other categories of FDA-reviewed products (you can read more about them here). In addition, Congress directed the FDA to work with the industry to set performance goals and create a process improvement plan, which you can read about here. Notably, the agency must deliver regular MDUFA IV performance reports to Congress, so that’s where I looked next to confirm my results.
The annual MDUFA performance reports are available on this website. Quarterly reports are available on a separate website. For both report types, results are reported for submissions within each FY as a cohort (e.g., Cohort 2021 is for FDA FY 2021, from October 2020 – September 2021). Since decisions often take more than a year or pass the FY boundaries, the cohorts aren’t complete until a few years after the FY ends. Data for incomplete cohorts are reported as preliminary in subsequent reports until the cohort is finished. The reports outline the program goals for different submissions in terms of the percent of submissions meeting a specified Review-Time goal. For example, under MDUFA IV, 95% of 510(k)s should have a Substantive Interaction within 60 calendar days and a Decision within 90 FDA days. 95% of Original PMAs should have a Substantive Interaction within 90 calendar days, with 90% receiving a decision in 180 FDA days with no Advisory Committee input. When Advisory Committee feedback is needed, the decision time is within 320 FDA days.
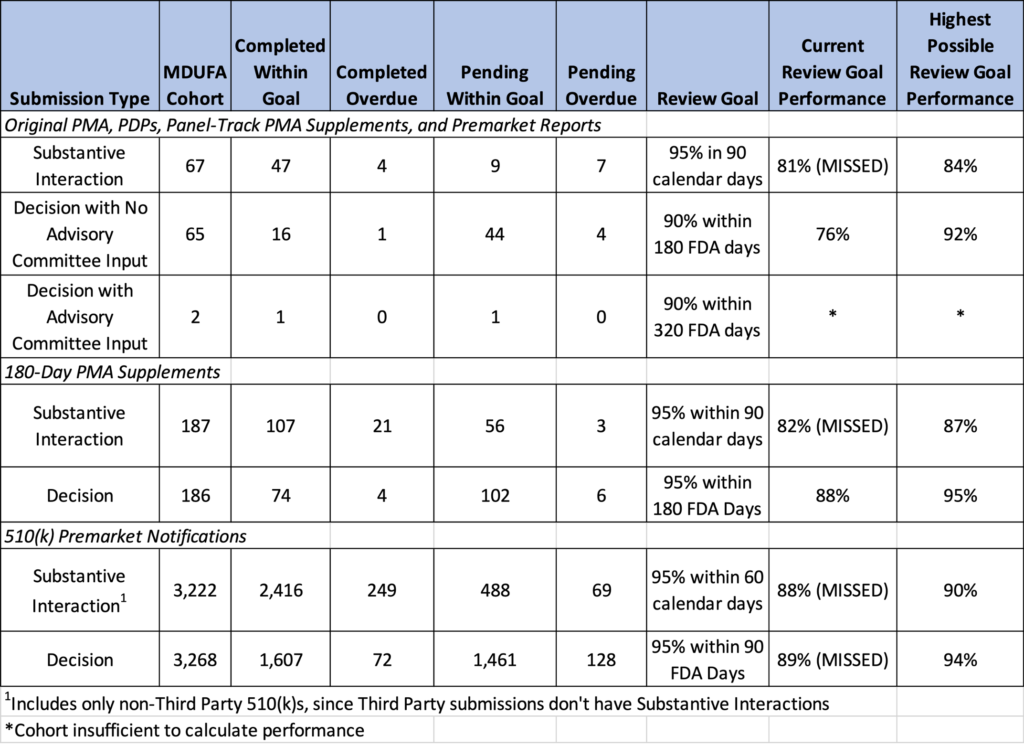
What does the FY2021 report say about the review performance for our target submissions? Figure 3 shows selected data from the FY2021 preliminary cohort results, which I adapted for this article. This table reveals an Original PMA has an 8 in 10 chance of a Substantial Interaction within 60 calendar days and a separate 8 in 10 chance of a decision within 180 FDA days. FDA days refer to the calendar days when the submission is under review, starting from the date an acceptable submission or amendment to make the submission acceptable is received. Together these metrics point to a 6 in 10 chance of an Original PMA decision within 240 days. Let’s also note that there’s a 4 in 10 chance the decision will take longer than 240 days (8 months). For 510(k)s, the stats are better, and the chances of a decision within 150 days are 8 in 10. The results from this MDUFA report are consistent with the historical data above. Assuming your submission is complete and immediately acceptable, these are reasonable estimates for your go-to-market milestones.
The 3Q FY2022 MDUFA report gave me the answer about success rates and other exciting information. Figure 4 is a copy of the graph showing year-over-year success rates for PMAs, while Figure 5 shows the diagram of success rates for 510(k)s. While PMA approval may take some time, the likelihood of success is high, with an 8 in 10 chance of approval on average over the last five years. Rates of Substantially Equivalent (SE) decisions for 510(k) submissions are similar, if not higher. Therefore, regardless of your submission pathway, your go-to-market estimates should plan for a high success rate and incorporate enough fiscal runway for an extended time to decide.
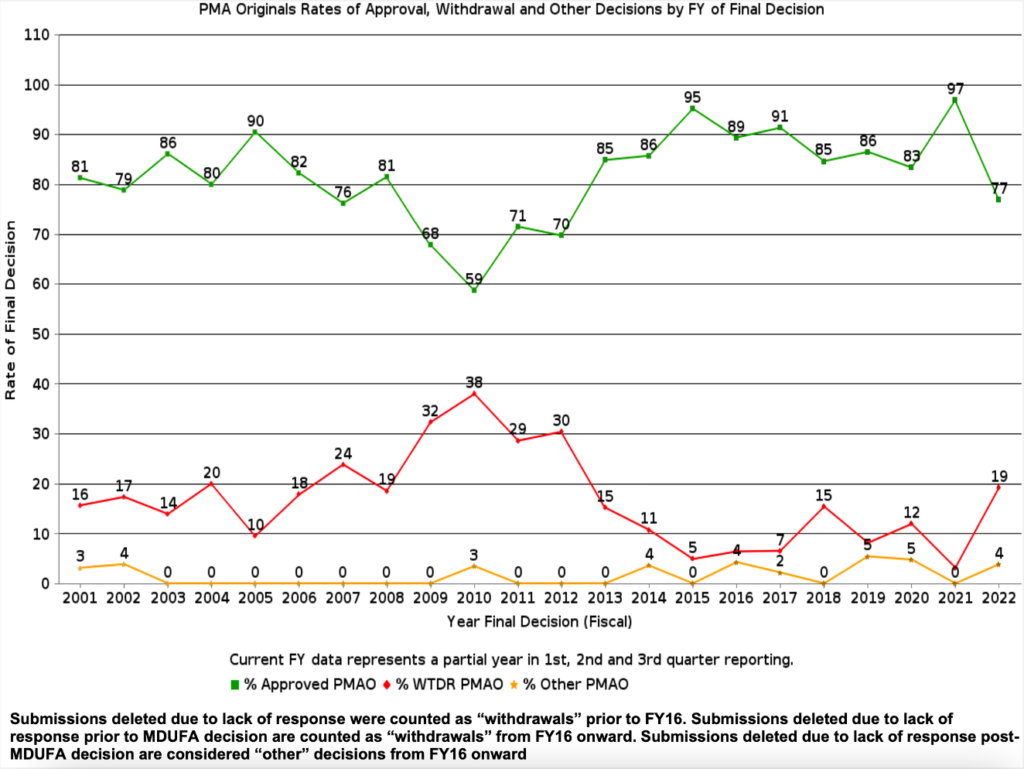
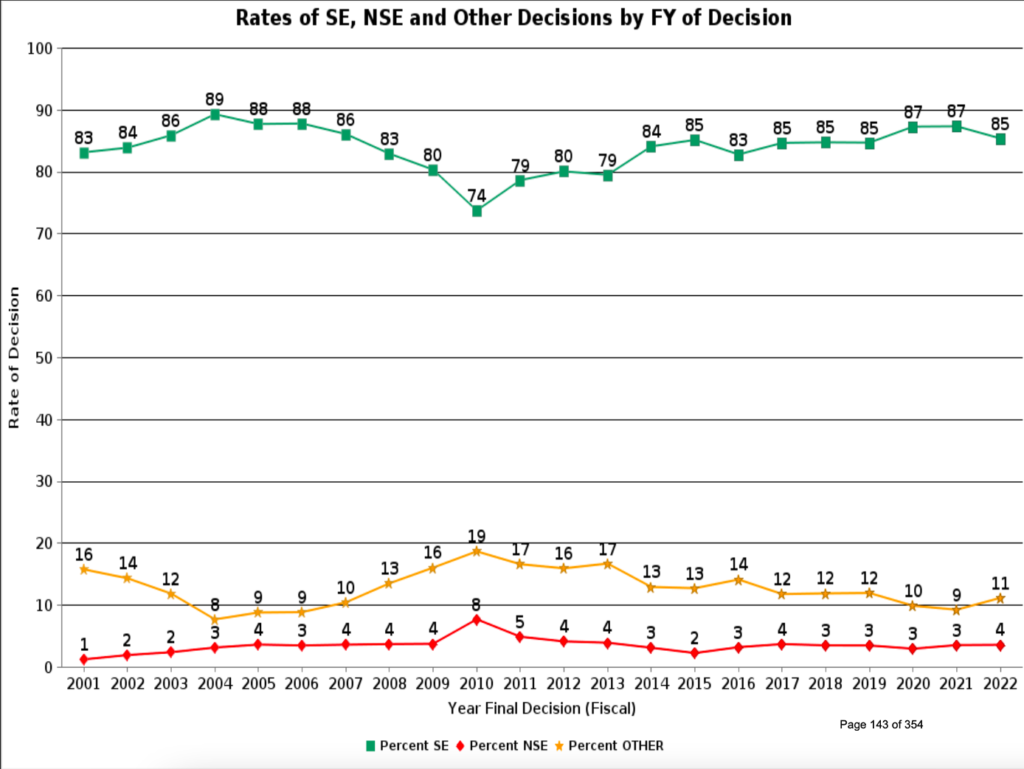
While the chances are good that your submission will receive a favorable decision, there is considerable variability in the time to receive agency approval. This variability has multiple causes. For example, reviewers found a major deficiency in over 80% of Original PMAs filed in 2021 (Figure 6). Similarly, 67% of 510(k)s required an additional information request (Figure 7). Therefore, avoid the pitfalls others have experienced with these four steps:
1. Plan to gather submission data in appropriate formats throughout development.
2. Use FDA information and programs to collect the correct data for your submission type.
3. Make sure your submission meets the preliminary administrative requirements. Appendix A of the 2021 MDUFA annual report contains a lookup table arranged by submission type.
4. Use the tools available to format your submission and include the required information. These include product-specific guidance checklists and eCopy templates.
Once you’ve filed your 510(k), keep an eye on your submission using the FDA’s progress tracker. The FDA will send your Official Correspondent an email notification if they are eligible for tracking access.
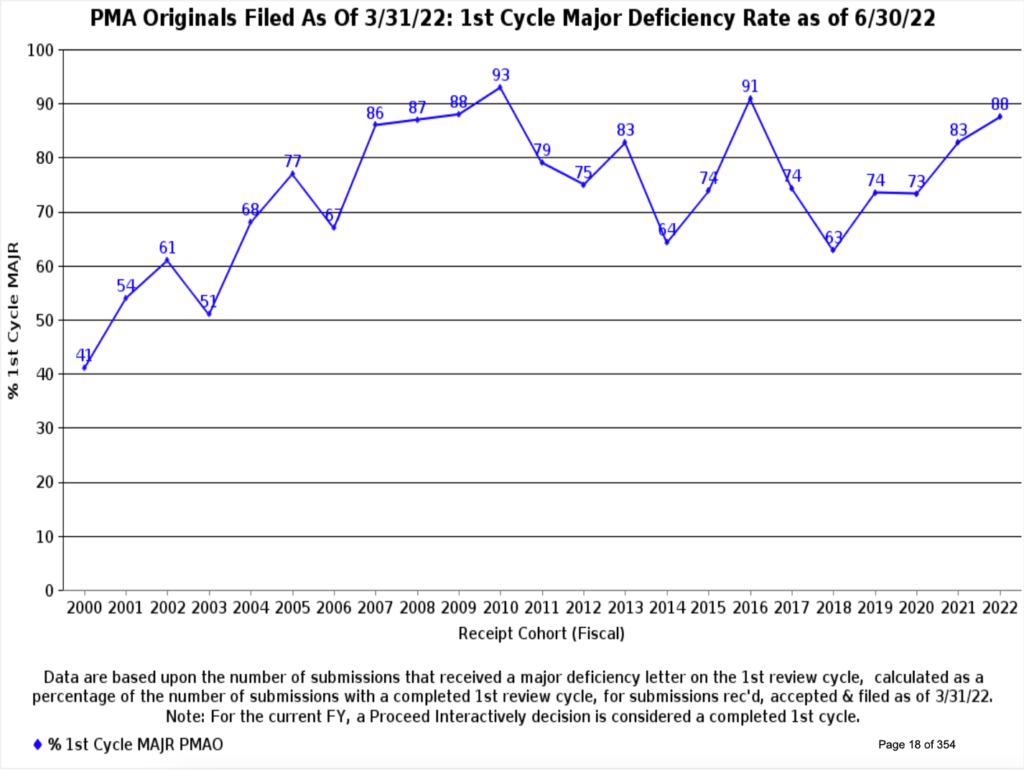
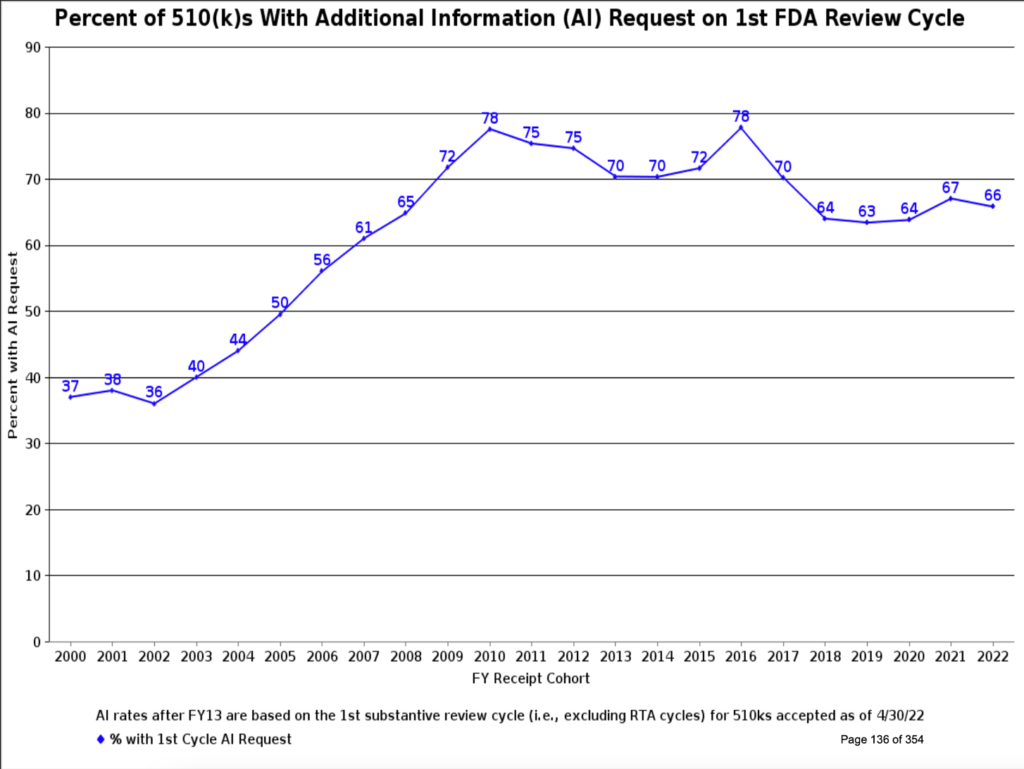
Readily available data from the FDA tells us we can expect a reasonably high (though not 100%) success rate for our PMA and 510(k) medical device submissions. The timeline for decisions can be highly variable and is undoubtedly affected by our planning and care in preparing the submission. MDUFA IV appears to be improving the decision time statistics. This program is in review for renewal as MDUFA V, and this article contains a good summary of the program’s progress in Congress as of September 2022. If timely device submission review is vital to your business, you should consider writing your Senator to express your support for this program.
If you enjoyed this article and would like to read more by Katrina, sign up for her newsletter.